共沉淀法制备磷酸锰铁锂正极材料全攻略
作者:贾雨 陈慧 刘梦娜 赵曦明 屈龙
单位:重庆科技大学化学化工学院
引用本文:贾雨, 陈慧, 刘梦娜, 等. 共沉淀法可控制备磷酸锰铁锂正极材料研究进展[J]. 储能科学与技术, 2026, 15(2): 419-434.
DOI:10.19799/j.cnki.2095-4239.2025.0852
本文亮点:系统论述了工业化共沉淀法在可控制备LMFP材料中的研究进展, 重点探讨了以磷酸盐和草酸盐为代表的前驱体及其在实现原子尺度上的均匀混合与组分精确控制方面的显著优势; 进一步阐述了共沉淀法与浓度梯度设计、碳包覆与离子掺杂等改性策略相结合对提升LMFP正极材料电子/离子电导率与结构稳定性方面的协同作用。
摘 要 橄榄石型磷酸锰铁锂(LiMnxFe1-xPO4, LMFP)作为下一代锂离子电池(LIBs)用正极材料,具有高能量密度、高安全性、低成本等优点。但LMFP本征电子电导率低、Li+扩散慢以及Mn3+引发的Jahn-Teller效应等关键因素制约其大规模应用。本文系统论述了工业化共沉淀法在可控制备LMFP材料的研究进展,重点探讨了以磷酸盐和草酸盐为代表的前驱体在实现原子尺度上的均匀混合与组分精确控制方面的显著优势;进一步阐述了共沉淀法与改性策略(如浓度梯度设计、碳包覆与离子掺杂)相结合对提升LMFP正极材料电导率与结构稳定性方面的协同作用。最后,本文强调了共沉淀法为高性能LMFP材料的可控制备提供可行方案,未来研究需致力于反应机理研究、工艺参数优化与多种策略的系统性整合,从而推动其在大规模储能系统中的商业化应用。
关键词 锂离子电池(LIBs);磷酸锰铁锂(LiMnxFe1-xPO4);正极材料;共沉淀法
在全球能源转型深入发展的背景下,发展高效、稳定的电化学储能系统已成为平衡可再生能源间歇性供应与电网稳定需求的关键支撑。锂离子电池(LIBs)凭借其高能量密度与长循环寿命,已在便携式电子设备、电动汽车及大规模储能系统中占据主导地位。当前在商业领域得到广泛应用的正极材料主要涵盖层状结构的钴酸锂(LiCoO2)与镍钴锰三元氧化物(LiNixCoyMnzO2, NCM,其中x+y+z=1)、尖晶石型结构的锰酸锂(LiMn2O4, LMO)、橄榄石结构的磷酸铁锂(LiFePO4, LFP)。其中,LiCoO2虽具有较高工作电压,但由于钴的毒性以及地壳中的低丰度,制约了其大规模应用与进一步发展;NCM材料具有高能量密度,但其热稳定性差、循环寿命较短及成本较高;LMO具有优异的倍率性能,但因Jahn-Teller效应容易发生容量衰减,循环性能较差;LFP具有优异的安全性和循环稳定性,但又具有较低的工作电压(约3.4 V vs. Li/Li+)使得能量密度较低,应用受到限制。因而,开发高能量密度、高安全性、低成本的新型正极材料,是下一代储能系统研究的关键目标之一。
正极材料高能量密度的特点主要由高工作电压实现。橄榄石型磷酸锰铁锂(LiMnxFe1-xPO4, LMFP)正极材料在充放电过程中因为存在由Mn2+/Mn3+氧化还原电对提供的电压平台(4.1 V vs. Li/Li+),其能量密度相较LFP具有显著优势。同时,LMFP在安全性与结构稳定性方面与LFP相当,所以被视为潜在的高比能锂离子电池正极材料。但是LMFP的实际应用仍面临若干关键挑战:①LMFP所具有的橄榄石型结构[图1(a)]使其内部缺乏连续的Mn(Fe)O6共棱八面体网络。因此,Li+只能沿一维的b轴方向进行扩散,从而导致其本征电子电导率与Li+扩散系数均显著偏低,严重制约材料的倍率性能;②在八面体晶体场中,Mn 3d轨道会发生能级分裂,形成能量较低的t2g轨道和能量较高的eg轨道[例如3dz2和3d(x2-y 2)轨道][图1(b)]。当eg轨道中存在单电子时(如Mn3+),这种不均匀的电子占据会打破八面体结构的对称性,导致Mn—O键发生畸变,即Mn3+的Jahn-Teller效应,该效应是导致材料发生晶格畸变、结构退化,并最终引起容量衰减与循环性能下降的主要原因。已有研究证实,即便在优化条件下,富锰LMFP(如Mn比例≥0.6)仍因相变应变与界面副反应存在显著的性能衰减,其容量保持率普遍较低。因此,针对上述挑战,通过合成工艺的优化实现对LMFP材料关键微观结构(如锰价态、颗粒形貌)的精准调控,已成为抑制Jahn-Teller效应、增强离子与电子传输能力的关键途径。
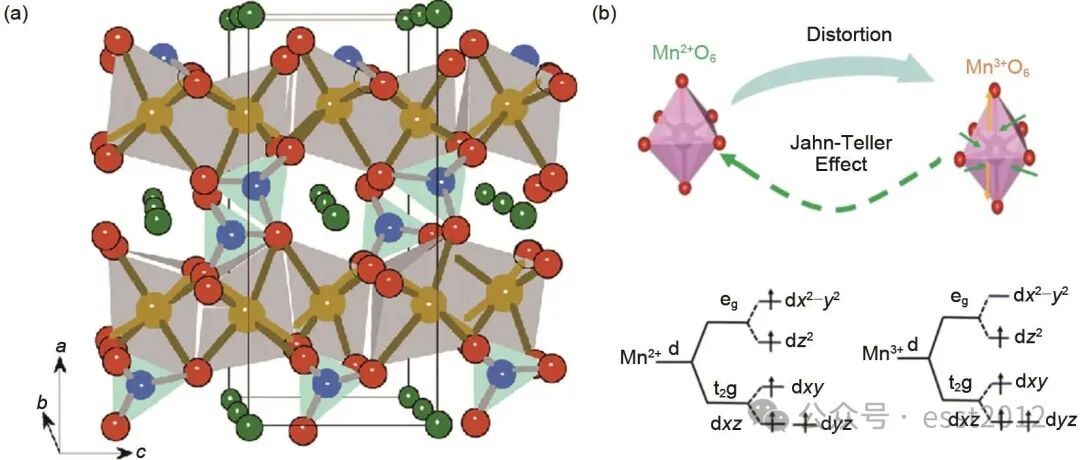
图1 (a) LiMn1-xFexPO4的晶体结构示意图;(b) Mn3+引起的Jahn-Teller畸变示意图
选择合适的制备方法对于提升LMFP的电化学性能以及实现其工业化生产至关重要。理论上,液相法(如溶胶-凝胶法、水热/溶剂热法和共沉淀法)更有利于实现Mn、Fe元素在原子尺度上的均匀分布。溶胶-凝胶法常通过络合剂螯合金属离子确保原子级均匀分布。随后,溶胶经凝胶化转变为固态前驱体,再经高温煅烧结晶化最终制备出LMFP。该方法具有工艺流程较为简便、操作步骤少、易于在实验室精确控制等优点,但其干燥过程较为复杂涉及有毒溶剂的使用,并且对设备的要求较高,这些因素制约其工业化应用。水热/溶剂热法的主要优势是可通过表面活性剂或模板剂调控产物的形貌与粒径。其典型工艺流程如下:将原料分散于水或有机溶剂中,在高压反应釜中进行热处理后经过烘干和煅烧步骤,合成出具有小粒径和高纯度的LMFP。但又具有设备成本高与操作条件复杂等缺点。此外,反应过程中需加入过量的锂源,尽管锂源可通过回收实现循环利用,但仍会显著增加制造成本。因此该方法的工业化应用仍面临较大挑战。
共沉淀法制备LMFP首先将可溶性锰盐与铁盐共溶解,随后加入沉淀剂,并通过精确控制反应条件(如pH值与温度)得到元素分布均匀的前驱体。随后,该前驱体经过离心、洗涤和干燥处理,再与锂源均匀混合通过高温煅烧制备出LMFP材料。共沉淀法具有能耗低、设备依赖性低,以及所得产物粒径小等优点,展现出良好的工业化应用前景。例如,Vanaphuti等利用可规模化生产的共沉淀法开发了一种无须使用氨、具有空气稳定性的新型前驱体,采用该前驱体制备的LMFP正极展现出优异的循环稳定性,在C/3倍率下经150次循环后容量保持率超过95%。
综上,采用共沉淀法可控制备前驱体结合固相高温煅烧工艺,可实现大规模制备高性能LMFP正极材料。通过系统调控反应物浓度、pH值及温度等关键参数,可制备多种磷酸盐与草酸盐前驱体,从而有效抑制分步沉淀与杂相的生成。此外,核壳/浓度梯度结构设计、碳包覆及离子掺杂等策略可与共沉淀法协同改性,进一步提升LMFP材料的电化学性能(图2)。例如,浓度梯度前驱体可有效抑制Jahn-Teller畸变,进而显著增强材料的循环稳定性;均匀的碳包覆层不仅能显著提升电子电导率,还能增强界面稳定性,从而使材料具有优异的倍率性能和长循环寿命;Mg2+、Nb5+等离子的掺杂可扩大Li+扩散通道、抑制晶格畸变并减少反位缺陷,从而有效增强材料的结构稳定性与离子传输动力学。因此,本文系统论述了共沉淀法制备LMFP前驱体的工艺路线,及其与上述多种改性策略的协同效应,旨在为高性能正极材料的开发提供理论依据与技术参考。
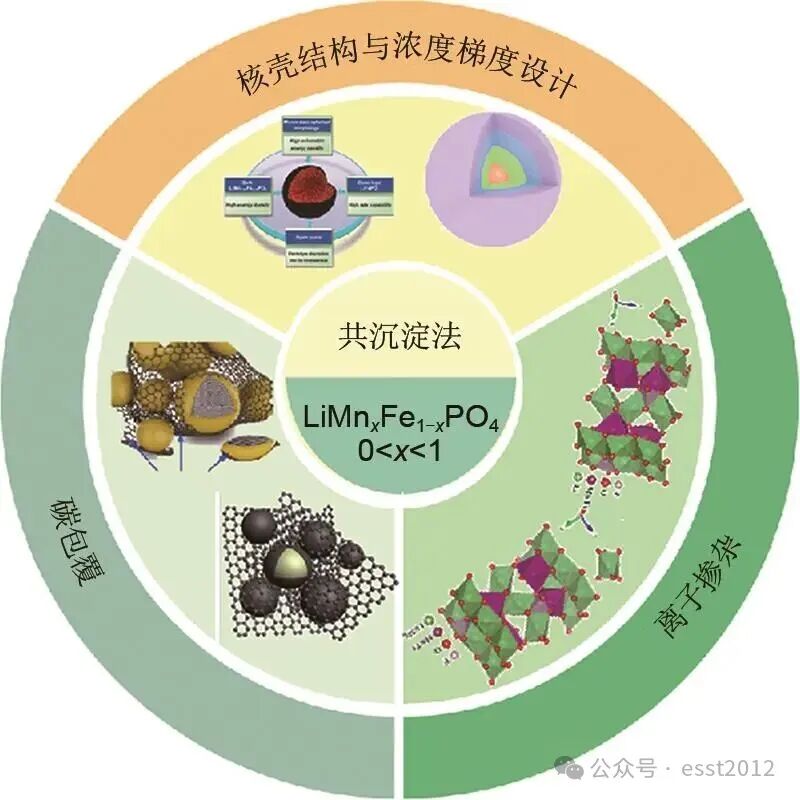
图2 共沉淀法制备磷酸锰铁锂改性策略
1 合理设计前驱体
LiMnxFe1-xPO4的化学组成为Li、Fe、Mn、P和O元素,锂元素通常由Li2CO3或LiH2PO4提供,并在高温煅烧阶段与前驱体混合,共沉淀法的目标主要是实现Fe、Mn及P元素的均匀分布,特别是在原子级尺度上抑制Fe与Mn的相分离并形成均匀固溶体。
共沉淀反应涉及成核与生长两个阶段。首先,溶液必须达到过饱和状态为过渡金属离子提供足够的化学势,从而才能克服成核能垒形成初始晶核,这是晶体生长的基础。随后,体系进入生长阶段,此时离子在晶核表面沉积,晶核通过奥斯特瓦尔德熟化(即小颗粒溶解并转移至大颗粒表面)进一步生长。根据成核机理,该过程可分为均相成核与异相成核:前者在均匀溶液体系中自发进行;后者则由外来杂质或晶种提供成核位点所诱导。这两种成核方式不同,其示意图如图3(a)所示。鉴于均相成核对于获得理想共沉淀产物的重要性,控制反应条件以促进均相成核是关键。
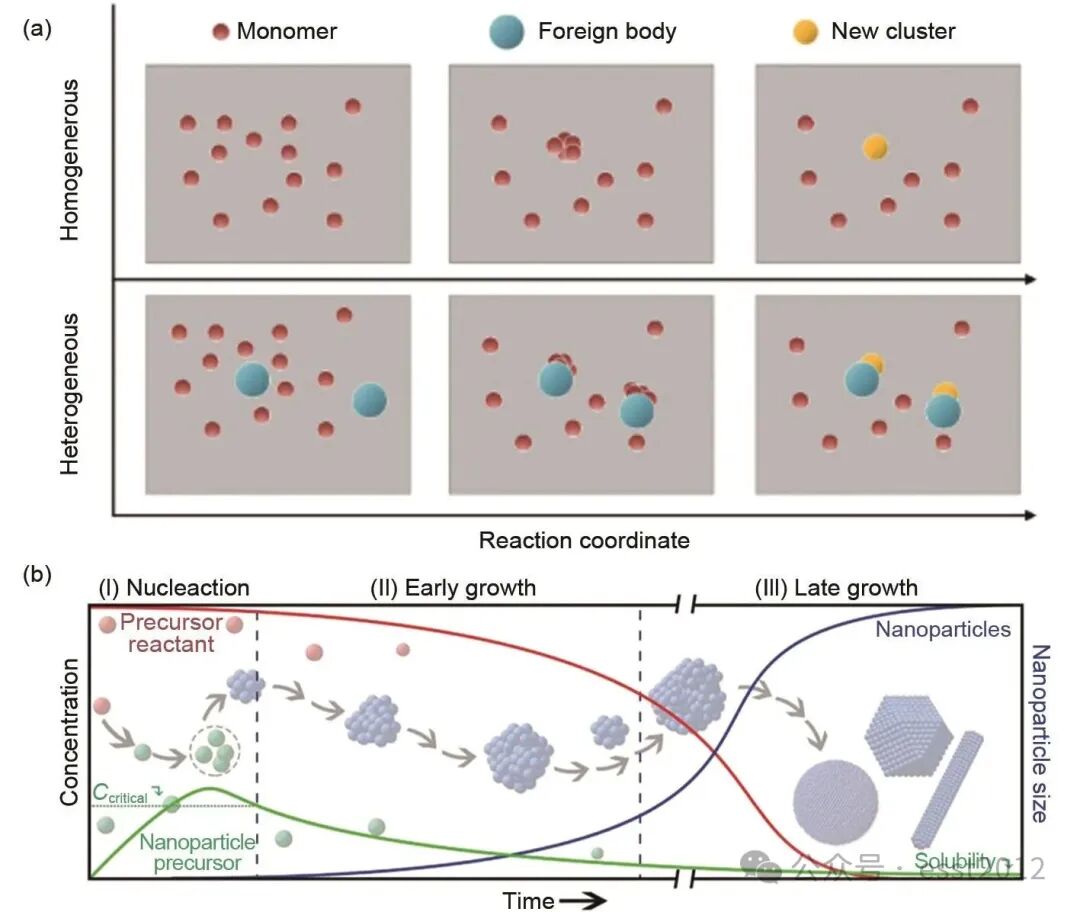
图3 (a) 均相成核与异相成核示意图;(b) Lamer模型示意图:反应物(红色)、反应物浓度(绿色)以及颗粒尺寸(蓝色)随时间依赖性演变的示意图,展示了溶液中晶粒形成的三个典型阶段。阶段I:过饱和度持续积累但未达临界成核浓度,无成核发生;阶段II:过饱和度超过临界值,引发爆发性均相成核,同时过饱和度迅速下降;阶段III:过饱和度降至临界值以下,成核停止,进入生长阶段
对于均相成核,其成核能垒(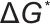 )与临界晶核半径(
)与临界晶核半径(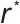 )可由经典成核理论(CNT)的核心公式描述:
)可由经典成核理论(CNT)的核心公式描述:
临界半径公式:
成核能垒公式: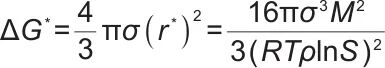
式中,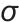 为界面张力;
为界面张力;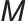 为溶质分子质量;
为溶质分子质量;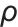 为溶质颗粒密度;
为溶质颗粒密度;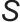 为过饱和度。
为过饱和度。
成核后,晶核生长速率成为关键参数,即单位时间内单位体积中形成的晶粒数,可描述为:
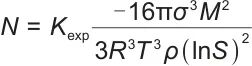 |
式中,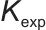 为反应速率常数。
为反应速率常数。
由以上两个式子可知,过饱和度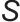 是成核与生长过程的核心。提高过饱和度会显著降低成核能垒
是成核与生长过程的核心。提高过饱和度会显著降低成核能垒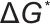 并减小临界半径
并减小临界半径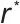 ,从而促进成核,产生数量多、尺寸小的颗粒;而降低过饱和度则有利于生长,使颗粒数量减少、尺寸增大。这一动力学过程可由经典的LaMer模型合理解释,如图3(b)所示,该模型描述了过饱和度随时间演变及其对成核与生长阶段的划分。pH通过改变目标沉淀物的溶解度来直接影响过饱和度
,从而促进成核,产生数量多、尺寸小的颗粒;而降低过饱和度则有利于生长,使颗粒数量减少、尺寸增大。这一动力学过程可由经典的LaMer模型合理解释,如图3(b)所示,该模型描述了过饱和度随时间演变及其对成核与生长阶段的划分。pH通过改变目标沉淀物的溶解度来直接影响过饱和度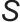 。为实现所有金属组分同步沉淀以形成均匀混合物,需快速将溶液pH调节至所有目标组分溶解度均极低的范围内,从而使所有金属离子同时达到高过饱和度并发生爆发性成核。为了获得小而均匀的纳米颗粒,需创造极高的瞬时过饱和度,快速、同步地完成成核过程。
。为实现所有金属组分同步沉淀以形成均匀混合物,需快速将溶液pH调节至所有目标组分溶解度均极低的范围内,从而使所有金属离子同时达到高过饱和度并发生爆发性成核。为了获得小而均匀的纳米颗粒,需创造极高的瞬时过饱和度,快速、同步地完成成核过程。
这一系列由过饱和度驱动的动力学行为,根本上是由体系内在的热力学平衡所决定。共沉淀反应本质上是多种金属离子在沉淀剂作用下,经历沉淀、络合、水解等多种反应动态耦合的化学平衡体系。这些反应最终驱使体系达到动态平衡,并遵循质量守恒、电荷守恒及物料守恒等热力学基本原则。因此,体系的过饱和度、pH值等关键状态变量均可通过分析平衡常数与组分浓度进行热力学估算,从而为理解与调控纯相前驱体的共沉淀过程提供理论依据。
热力学平衡是实现高效沉淀反应与精确化学计量控制的基础。以Mn2+-Fe2+-Mg2+-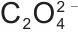 -H2O体系为例,其构建的热力学模型,如图4(a)~(d)所示,旨在通过对宏观反应条件的精确调控,促使体系发生均匀成核而非分步沉淀,从而在分子/原子尺度上实现金属离子的同步沉积。采用草酸盐沉淀M2+金属离子时,存在一个最优的pH值与沉淀剂用量范围。沉淀剂过量系数不足或pH过低时,溶液的过饱和度不足,这不仅导致金属离子沉淀不完全,还会引发非均相成核或在现有晶核上的缓慢生长,从而造成元素偏析;反之,pH过高或过量系数过大时,草酸根会由沉淀剂转变为络合剂,与金属离子形成可溶性络合物[如
-H2O体系为例,其构建的热力学模型,如图4(a)~(d)所示,旨在通过对宏观反应条件的精确调控,促使体系发生均匀成核而非分步沉淀,从而在分子/原子尺度上实现金属离子的同步沉积。采用草酸盐沉淀M2+金属离子时,存在一个最优的pH值与沉淀剂用量范围。沉淀剂过量系数不足或pH过低时,溶液的过饱和度不足,这不仅导致金属离子沉淀不完全,还会引发非均相成核或在现有晶核上的缓慢生长,从而造成元素偏析;反之,pH过高或过量系数过大时,草酸根会由沉淀剂转变为络合剂,与金属离子形成可溶性络合物[如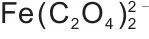 ,
, 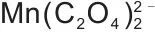 ],这会显著降低溶液的有效过饱和度,抑制成核,同样不利于均匀沉淀,并导致沉淀效率下降。通过对各金属离子沉淀行为的最优参数范围进行交叉分析,图4(d)确定了一个优化的工艺窗口:将草酸盐过量系数控制在1.10,反应pH值维持在2.0~3.5。在该条件下,溶液中所有金属离子(Fe2+、Mn2+和Mg2+)的浓度积同时且快速地超过其草酸盐沉淀的临界过饱和度,从而实现高效且稳定的共沉淀。这一状态触发了“爆发式均匀成核”。大量晶核在瞬间同时生成,迅速消耗溶液中的自由金属离子,使得体系中各点的成分被“冻结”在初始的、均匀的溶液状态,从而有效避免因各离子沉淀溶度积(Ksp)差异而可能导致的分步沉淀。因此,通过精确调控实验参数,可在分子水平上实现金属离子的共沉淀,从而有效防止分步沉淀。图4(e)展示的EDS图谱证明了Fe与Mn的均匀分布。
],这会显著降低溶液的有效过饱和度,抑制成核,同样不利于均匀沉淀,并导致沉淀效率下降。通过对各金属离子沉淀行为的最优参数范围进行交叉分析,图4(d)确定了一个优化的工艺窗口:将草酸盐过量系数控制在1.10,反应pH值维持在2.0~3.5。在该条件下,溶液中所有金属离子(Fe2+、Mn2+和Mg2+)的浓度积同时且快速地超过其草酸盐沉淀的临界过饱和度,从而实现高效且稳定的共沉淀。这一状态触发了“爆发式均匀成核”。大量晶核在瞬间同时生成,迅速消耗溶液中的自由金属离子,使得体系中各点的成分被“冻结”在初始的、均匀的溶液状态,从而有效避免因各离子沉淀溶度积(Ksp)差异而可能导致的分步沉淀。因此,通过精确调控实验参数,可在分子水平上实现金属离子的共沉淀,从而有效防止分步沉淀。图4(e)展示的EDS图谱证明了Fe与Mn的均匀分布。
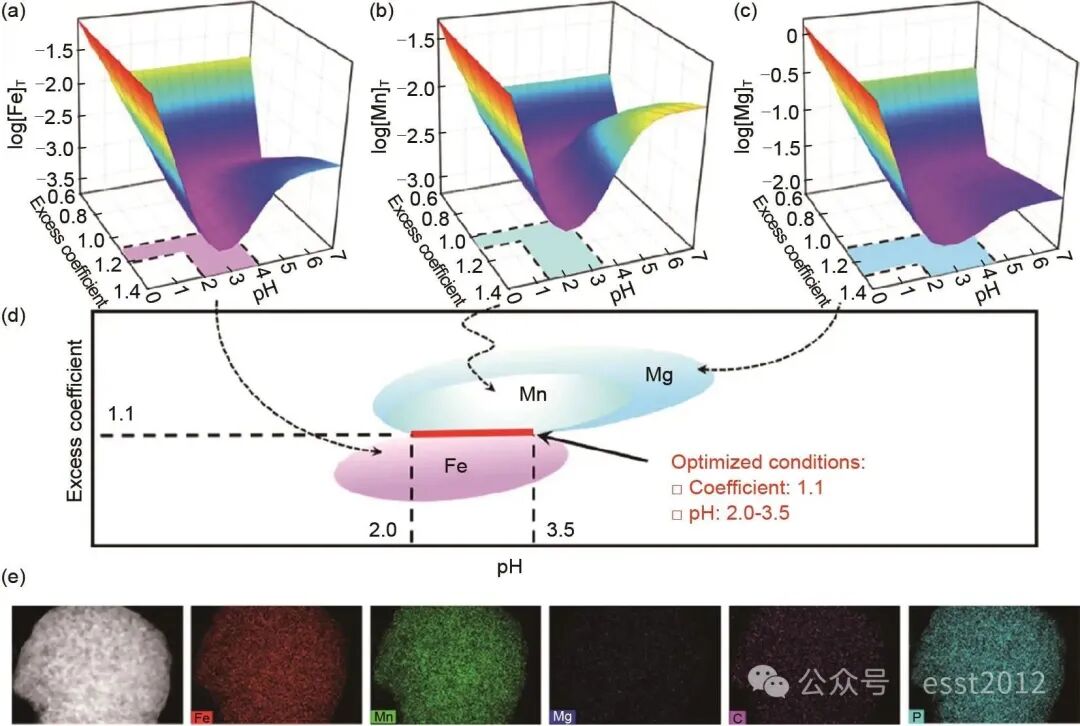
图4 Mn2+-Fe2+-Mg2+-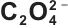 -H2O体系的热力学平衡分析:(a)~(c) 草酸盐过量系数、pH与金属总浓度对数三维关系图;(d) 沉淀条件优化结果;(e) Li(Fe0.4Mn0.6)0.97Mg0.03PO4/C的EDS图
-H2O体系的热力学平衡分析:(a)~(c) 草酸盐过量系数、pH与金属总浓度对数三维关系图;(d) 沉淀条件优化结果;(e) Li(Fe0.4Mn0.6)0.97Mg0.03PO4/C的EDS图
根据沉淀剂的不同,沉淀体系主要分为两类,包括磷酸盐类[如MnxFe1-xPO4、(MnxFe1-x)3(PO4)2和NH4MnxFe1-xPO4]以及草酸盐[如MnₓFe1-xC2O4]。精确调控前驱体的特性(如形态与尺寸)并优化热处理参数,是制备高性能LMFP正极材料的关键策略。
磷酸盐前驱体通过预先引入磷元素并构筑PO4四面体框架,能有效抑制锂插入过程中的结构重排,进而促进橄榄石结构的形成。早期研究主要采用MnxFe1-xPO4作为前驱体。通常将原料与H3PO4混合并且需要加入氧化剂(如HNO3、H2O2),金属离子经过氧化,得到三价的磷酸锰铁前驱体,但是由于Mn3+离子在水溶液中热力学稳定性较差,在pH<2的条件下才能稳定存在,并且需要使用有机溶剂降低溶液的介电常数,加速离子间的结合反应,提高成核速率。因此,此类前驱体的合成须在有机溶剂(如乙醇)中进行,而且在后续煅烧过程中需要H2或还原碳的引入。这不仅增加了生产成本,还引发了环境顾虑,严重限制LMFP正极材料的大规模产业化应用。
研究者的重心转移至利用在水溶液中稳定的Mn2+来合成(MnxFe1-x)3(PO4)2。该前驱体反应条件更温和,以MnSO4、FeSO4等为廉价原料,NH4H2PO4作为沉淀剂,在N2保护气氛下通过氨水调节溶液pH≈6.5进行反应,生成(MnxFe1-x)3(PO4)2沉淀,通常为微米级片状或疏松纳米片堆叠结构;后续喷雾干燥可形成粒径为1~4 μm的均匀球形二次颗粒,该方法不仅更具环境友好性,并且在后续的锂化过程中无须添加还原碳。例如,Wang等提出了一种原子经济性高的策略,合成了单斜结构的(MnxFe1-x)3(PO4)2/GO前驱体。该方法不仅避免了传统生产过程中产生的含重金属废水,而且GO的加入抑制颗粒生长,提供成核位点,促进元素均匀分布,而且经过烧结后,直接得到了被三维导电网络包覆的LMFP/rGO/C复合材料。该复合材料表现出优异的电化学性能[图5(b)]:在0.05、0.2、0.5、1、2和5 C倍率下,其放电容量分别为156.4、152.3、148.2、142.2、136.2和127.5 mAh/g。
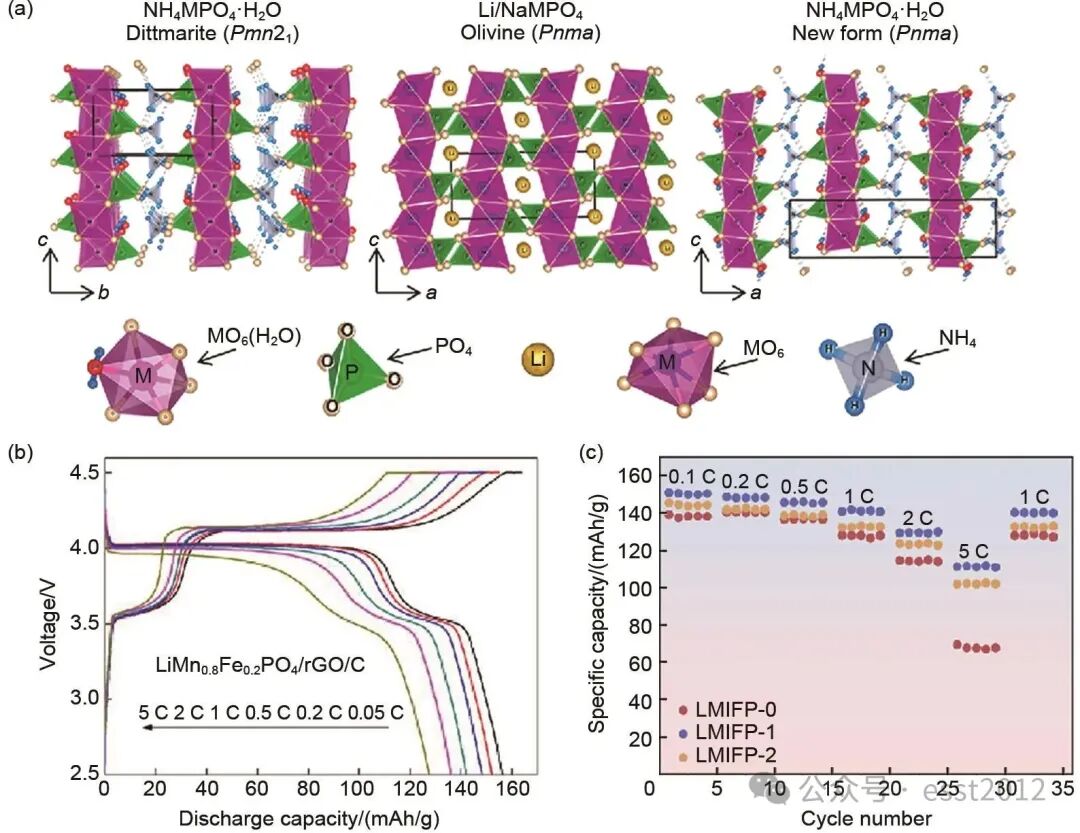
图5 (a) NH4MPO4·H2O和LiMPO4的晶体结构,以及M-PO4层的排列方式;(b) LiMn0.8Fe0.2PO4/rGO/C复合材料在不同充放电倍率下的充放电曲线;(c) LiMn0.5978Fe0.3522Mg0.0506PO4/C正极材料倍率性能
以MnxFe1-xPO4或(MnxFe1-x)3(PO4)2为前驱体合成LiMnxFe1-xPO4时,其锂化过程伴随晶型转变与结构重排,导致最终获得的LMFP产物在形貌和结构上与理论存在较大偏差。为解决上述问题,研究人员提出采用结构与橄榄石相似的迪特马矿(Dittmarite)NH4MPO4·H2O作为前驱体。如图5(a)所示,迪特马矿与橄榄石具有相似的晶体框架,均由MO6八面体和分布于ac与bc平面的PO4四面体构成。这种结构相似性极大地促进了NH4+与Li+的离子交换反应,从而高效地驱动该前驱体向橄榄石型LMFP的转化。因此,凭借其优异的结构相容性与转化动力学优势,NH4MPO4·H2O被视为是一种理想的前驱体。该前驱体制备过程通常是将(NH4)2HPO4溶液与含MnSO4·H2O和(NH4)2Fe(SO4)2·6H2O的硫酸盐溶液按Mn/Fe比例混合,在70~75℃下搅拌反应,冷却后于母液中老化2天,产物经过滤、水洗、酒精洗涤后室温干燥。Wu等采用NH4Mn0.6Fe0.4PO4·H2O作为前驱体,实现了Mn、Fe、P元素的原子级均匀共沉淀。所制备的正极材料展现出优异的倍率性能[图5(c)],其在0.1、1和5 C倍率下的放电容量分别达到150.1、141.4和111.6 mAh/g。
草酸盐前驱体的合成主要涉及金属盐(如FeSO4、MnSO4或其乙酸盐)与草酸根在水溶液中的反应,以生成过渡金属草酸盐。合成过程中常使用过量的草酸根或氨水以调控pH至近中性(约4.5),并加入添加剂如抗坏血酸来抑制氧化。沉淀剂的选择和反应温度对共沉淀过程具有显著影响。例如,Park等采用草酸盐共沉淀法合成多组分的Mn1/3Fe1/3Co1/3(C2O4)·2H2O前驱体。该合成路径通过精确控制pH值、温度等关键参数,有效解决了低pH下锰溶解和高pH下铁氧化的矛盾。具体而言,当使用草酸与过渡金属硫酸盐制备前驱体时,由于草酸锰溶解度较高,产物中会出现明显的锰缺失现象;而改用草酸铵作为草酸盐源可提高溶液pH值,从而使产物更接近目标化学计量比。在室温共沉淀时,铁倾向于形成β相草酸盐,而在90℃共沉淀时则生成α相草酸盐;锰在所有条件下均仅形成α相草酸盐。为获得相纯且多组分均匀混合的前驱体,在草酸盐体系中需将温度维持在90℃,以防止富锰α相与贫锰β相草酸盐的相分离。
与磷酸盐共沉淀相比,草酸盐共沉淀具有以下优势:Mn、Fe、Ni、Co以及Mg的草酸盐溶度积常数(Ksp)相近,这样能够形成稳定的草酸二水合物,并实现多种金属离子同步析出,该特性可促进共沉淀过程的同步性与均匀性,从而获得化学成分高度均匀的前驱体,并且可以在共沉淀过程中进行原位掺杂,更有利于实现掺杂离子的原子级均匀分布。因此,该方法能够精确控制Mn/Fe比例,并确保元素在晶体结构中的均匀分布。这些因素直接影响最终材料的能量密度、倍率性能与循环稳定性。草酸盐前驱体因其Mn/Fe比例可在较宽范围内连续调节、分解温度低于锂化温度,且在分解过程中体积变化较小、无残留杂质,因而被视为一种理想的前驱体体系。例如,Wang等采用草酸钠缓冲溶液作为沉淀剂,有效提升沉淀效率,同时维持pH恒定,成功合成了单斜α相结构的Mn0.5Fe0.5C2O4·2H2O前驱体,并在合成过程中加入抗坏血酸用于抑制金属离子氧化[图6(a)]。与传统固相法相比,采用共沉淀法制备的前驱体所合成的正极材料,呈现出更利于锂离子扩散的晶体结构,从而显著提升了材料的倍率性能[图6(b)]。该材料在10 C的高倍率下,放电容量仍达到108.6 mAh/g。
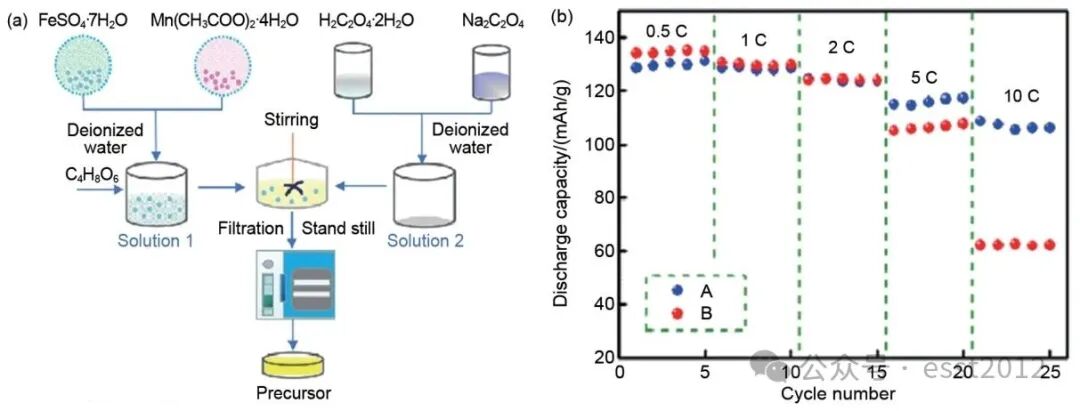
图6 (a) Mn0.5Fe0.5C2O4·2H2O前驱体合成示意图;(b) LiMn0.5Fe0.5PO4/C倍率性能图
总之,LiMnxFe1-xPO4的合成通常基于两类前驱体:磷酸盐与草酸盐。如表1所示,通过对未改性基础材料的系统对比,不同前驱体的选择在合成路径、产物特性及最终电化学性能上呈现出较大差异。在磷酸盐体系中,MnxFe1-xPO4的合成条件较为苛刻,且放电容量较低(0.1 C下>140 mAh/g)。而(MnxFe1-x)3(PO4)2的合成则相对简易,同MnxFe1-xPO4相比放电容量有较大提升(0.1 C下>150 mAh/g),但其锂化过程会引发显著的结构重排,从而导致前驱体的形状与尺寸难以维持,这制约其电化学性能的进一步提升,使其倍率性能(如5 C容量约110 mAh/g)仍存在提升空间。相比之下,NH4MnxFe1-xPO4的合成难度较低,凭借与橄榄石结构的高度相似性,实现低应变的结构转化,在其锂化过程中未观察到显著的结构重排,从而在获得良好容量(0.1 C下约145 mAh/g)的同时,展现出了更优的倍率性能(5 C下约120 mAh/g)。草酸盐前驱体不仅有利于实现均匀沉淀与精准的Mn/Fe比例控制,更是实现原子级均匀混合的有效体系,其制备的材料展现出高的放电容量与倍率性能(如0.1 C下>155 mAh/g,10 C高倍率下仍能保持100 mAh/g以上)。基于上述讨论,从合成工艺的可行性、结构转化的稳定性与最终电化学性能(特别是高倍率性能)三个维度综合评估,草酸盐前驱体(MnxFe1-xC2O4)与NH4MnxFe1-xPO4均可被视为制备LiMnxFe1-xPO4的理想前驱体。
表1 不同前驱体合成LiMnxFe1-xPO4/C的对比总结 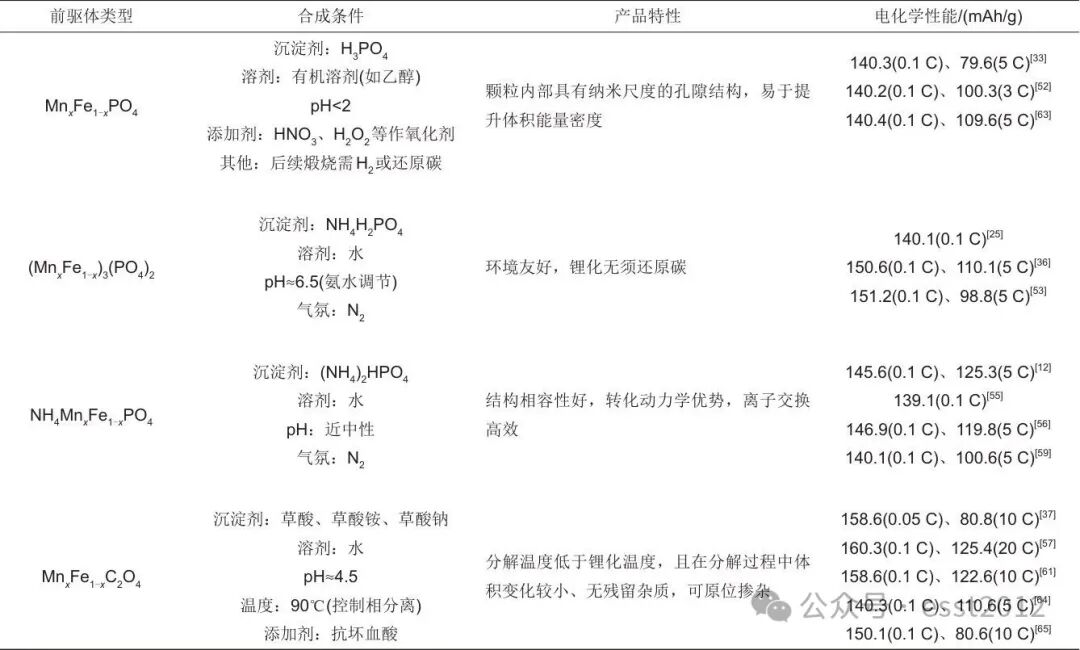
2 共沉淀法协同其他改性方法的策略
研究者通过外部调控手段优化共沉淀制备过程,以提升LMFP正极材料的综合性能,但是该材料仍存在较低的电子电导率、较低的Li+扩散系数以及Mn3+引发的Jahn-Teller效应等问题。因此,有必要将共沉淀法与其他改性策略相结合,以协同解决LMFP材料合成过程中无法解决的电化学性能局限性。目前,制备高性能LiMnxFe1-xPO4材料的常见改性策略主要包括核壳结构与浓度梯度设计、碳包覆及离子掺杂等。
2.1核壳结构和浓度梯度设计
共沉淀法基于其球形前驱体在连续搅拌釜式反应器中的可控生长行为,适用于制备具有核壳或浓度梯度结构的前驱体,该方法适用于正极材料的工业化规模生产,并在锂离子电池领域应用广泛。核壳结构作为一种广泛应用的设计策略,通过在正极材料中构建功能分区的异质界面,可协同提高体积能量密度与循环稳定性,从而改善材料的电化学与力学性能。例如,Oh等通过在LiMn0.85Fe0.15PO4内核表面包覆LiFePO4外壳,如图7(a)所示,制备了一种核壳型双相正极材料。该核壳结构电极展现出最高的充放电容量[图7(b)],约为152 mAh/g。在0.5 C倍率下,其放电比容量较未修饰材料显著提升[图7(c)],并表现出良好的循环稳定性。
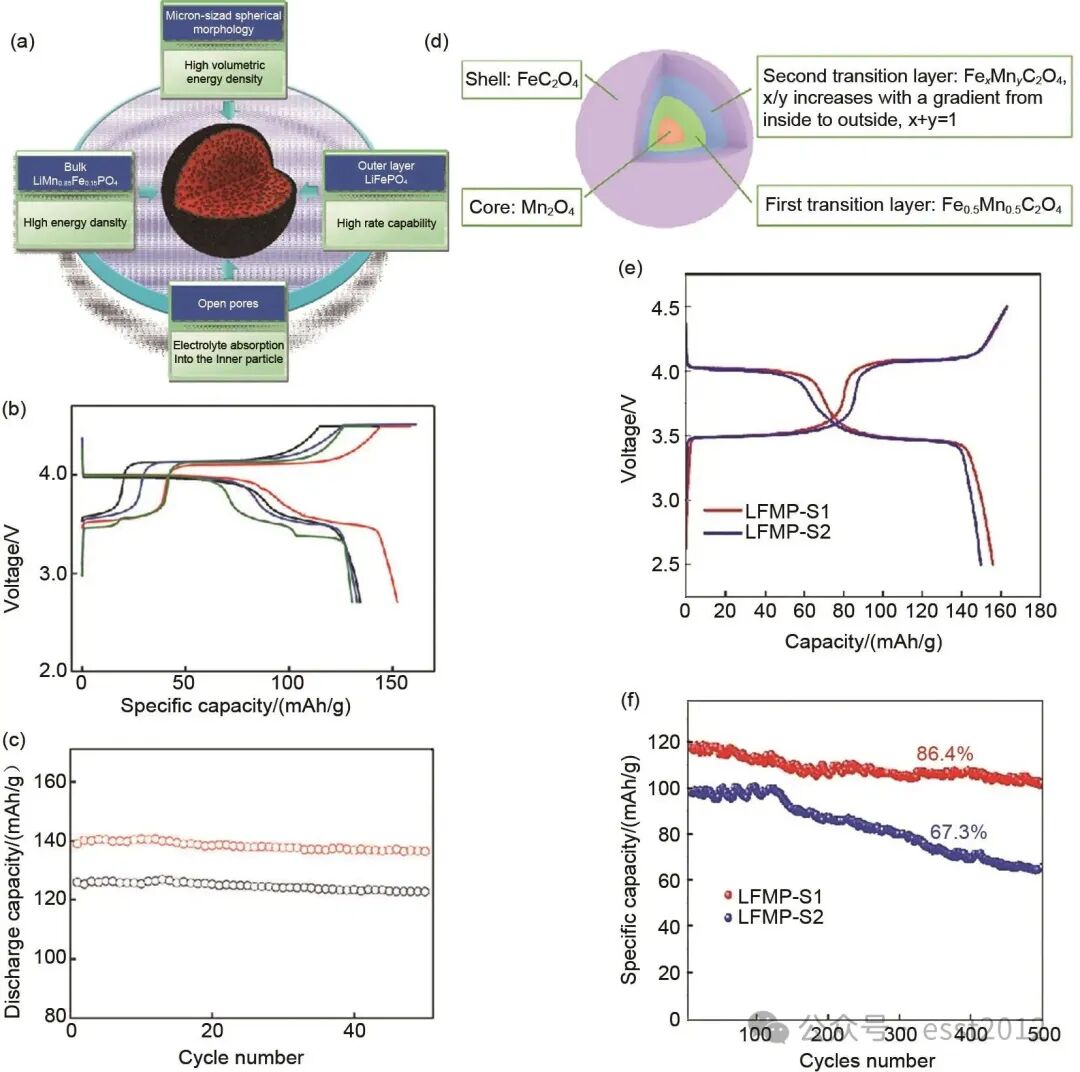
图7 (a) 核壳结构设计示意图;(b) LiMn0.85Fe0.15PO4/C、LiMn0.67Fe0.33PO4/C、LiMn0.65Fe0.35PO4/C(无核壳结构)、LiMn0.85Fe0.15PO4-LiFePO4/C(核壳结构)0.1 C倍率下的首次充放电曲线对比;(c) LiMn0.85Fe0.15PO4/C、LiMn0.85Fe0.15PO4-LiFePO4/C(核壳结构)在0.5 C倍率下的循环性能对比;(d) Mn0.5Fe0.5C2O4·2H2O前驱体示意图;(e) LiMn0.5Fe0.5PO4复合材料0.1 C倍率下的充放电曲线对比;(f) 1 C倍率下超长寿命循环性能对比
核壳结构在提高体积能量密度方面具有显著优势,而浓度梯度设计则通过调控元素分布,进一步缓解了Mn基橄榄石材料的界面不稳定性问题,从而显著提升了材料的循环寿命与倍率性能。Jiang等通过共沉淀法制备了浓度梯度前驱体Fe0.5Mn0.5C2O4·2H2O[图7(d)],Fe与Mn元素含量从颗粒中心向边缘呈现递减的梯度变化,且中心区域元素分布较为均匀。该梯度结构有效抑制了电极-电解液副反应与Mn3+溶解。这使得材料在0.1 C下的放电容量达到156 mAh/g[图7(e)],且在1 C倍率下经过500次循环后,容量保持率为86.4%(未改性材料为67.3%),显著改善了材料的循环稳定性[图7(f)]。
2.2碳包覆
LiMnxFe1-xPO4材料具有高理论容量和良好的安全性,但其实际应用仍面临本征电导率低、循环过程中颗粒易长大等问题的制约。碳包覆层可作为电子导体以增强材料的整体电导率,同时在合成过程中抑制颗粒过度生长,促进纳米结构的形成。碳包覆的效果取决于其均匀性、厚度及碳源类型,但过厚或不均匀的包覆层可能阻碍锂离子扩散。例如,Oh等采用双碳源包覆技术,合成了LiMn0.85Fe0.15PO4/C橄榄石材料。研究发现,碳层在颗粒孔隙及表面的分布形态因碳源种类不同而存在差异[图8(a)]。均匀的碳包覆层可有效提升材料的导电率,抗坏血酸-沥青(7.9×10-2 S/cm)混合碳源包覆的粉末比PVP-沥青(1.1×10-3 S/cm)混合碳源包覆的粉末具有更高的导电性。使用抗坏血酸-沥青混合碳源包覆的核壳电极展现出最优的电化学性能:其在0.05 C下的放电容量为154 mAh/g,在5 C倍率下仍可保持100 mAh/g。此外,Du等制备了LiMn0.8Fe0.2PO4/C材料,其碳层厚度为2~3 nm。该材料在10 C高倍率下仍能展现出82 mAh/g的容量。这一结果进一步证实,碳包覆层的均匀性和厚度是影响其电化学性能的关键因素。
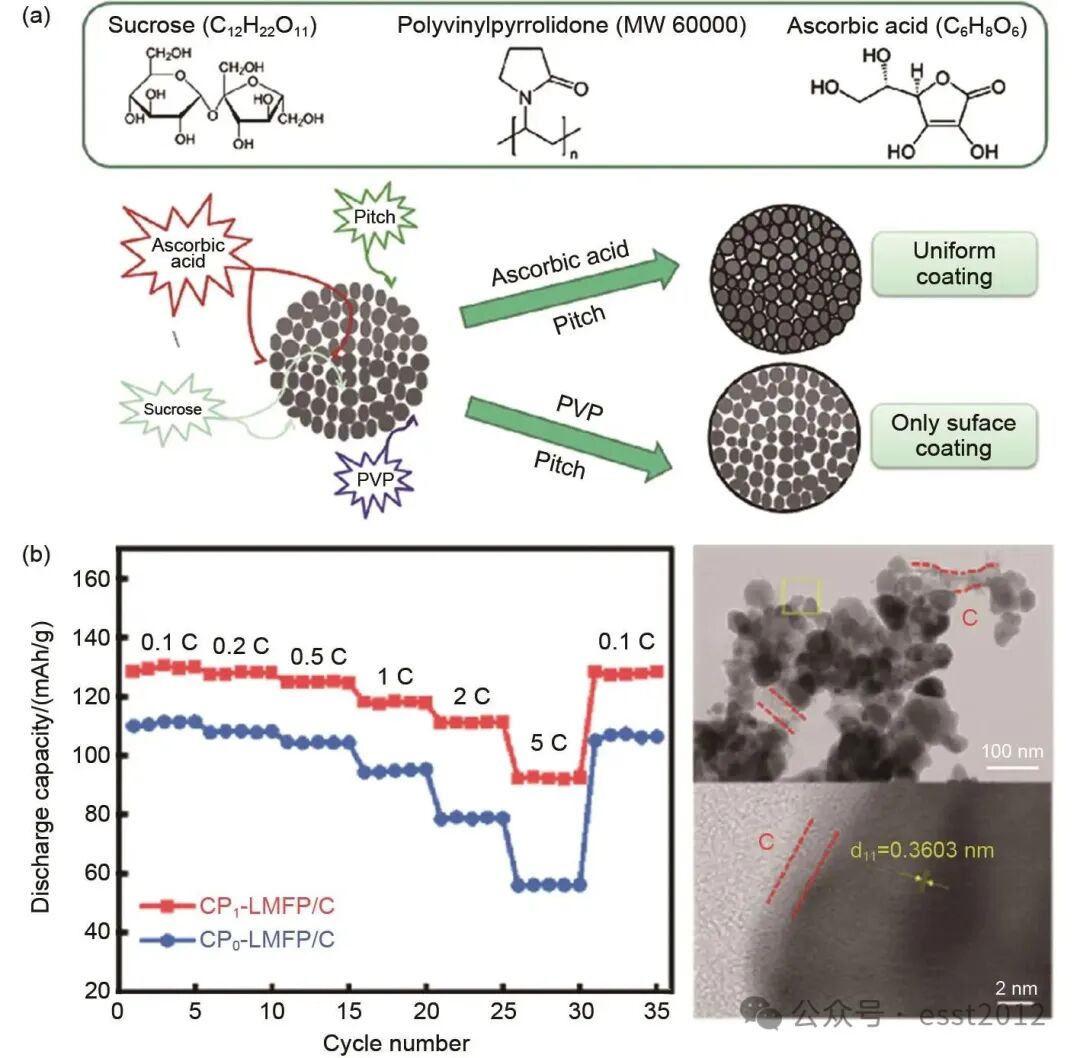
图8 (a) 不同碳源的碳包覆效果示意图;(b) CP1-LMFP/C和CP0-LMFP/C的倍率性能图、CP1-LMFP/C的TEM图像与相应的FFT点
尽管不同研究采用的合成方法与材料组成各不相同,但其结果一致表明,适量的碳包覆可有效抑制循环过程中的极化现象并减缓结构退化,抑制颗粒长大,从而增强材料的电化学稳定性。Wang等通过共沉淀法结合多段碳化热处理工艺,引入聚苯乙烯纳米球作为模板剂和碳源,制备了CP1-LiMn0.8Fe0.2PO4/C纳米复合材料。在热处理过程中,聚苯乙烯纳米球不仅可形成导电碳层、优化电子传输路径,还能细化颗粒尺寸并有效抑制纳米颗粒团聚。相互连通的导电碳网络显著提升了锂离子扩散系数,从而显著提高了LMFP材料的放电比容量和循环性能。如图8(b)所示,该CP1-LiMn0.8Fe0.2PO4/C材料表现出优异的倍率性能,在5 C高倍率下容量仍保持92.8 mAh/g。Fang等合成的LiFe0.67Mn0.33PO4/C材料在1 C倍率下循环200次后,容量保持率高达96.16%。这些结果共同凸显了碳包覆对增强材料倍率性能与循环稳定性的重要作用,但碳材料本身并非电化学活性物质,过量使用会降低材料的整体比容量,从而导致体积能量密度降低。因此,在实际应用中必须在导电性与活性物质含量之间寻求优化平衡。碳包覆通过增强界面稳定性和优化电荷传输动力学,成为提升电极材料高倍率性能与长循环寿命的一种有效策略。
2.3离子掺杂
离子掺杂作为一种关键改性手段,通过精确调控晶体结构调整Mn—O键和离子传输行为,有效抑制Jahn-Teller效应与Mn3+的溶出等现象。共沉淀法因其可实现掺杂离子的原子级均匀分布的优势,已成为优化正极材料结构一致性和电化学性能的重要合成策略。通过调控共沉淀过程的热力学参数,可实现多种金属离子同步析出,从而在前驱体合成阶段达到原子级均匀分布,该均匀性为后续高温固相合成成分一致的体相掺杂材料提供了保障。Xin等采用共沉淀法合成了NH4Mn0.6Fe0.4PO4·H2O前驱体,该前驱体经高温热处理后得到Nb掺杂的LMFP正极材料(LMFP/Nb@C),图9(a)展示了通过DFT计算研究的晶体结构,在LMFP/Nb@C中,Mn—O键长介于2.1109~2.2574 Å,畸变幅度为0.1465 Å。相比之下,未掺杂样品LMFP@C的Mn—O键长变化更大(2.0843~2.2692 Å),畸变值为0.1848 Å。研究表明,Nb5+掺杂通过调控电子结构,有效抑制了Jahn-Teller畸变,减轻了MnO6八面体的结构变形,优化了其局部配位环境,从而显著提升了晶体结构的整体稳定性。LMFP/Nb@C材料表现出优异的倍率性能,如图9(b)所示:在0.1 C、0.2 C、0.5 C、1 C、2 C和5 C倍率下,其放电比容量分别为155.63 mAh/g、154.69 mAh/g、151.54 mAh/g、148.29 mAh/g、144.03 mAh/g和134.00 mAh/g。
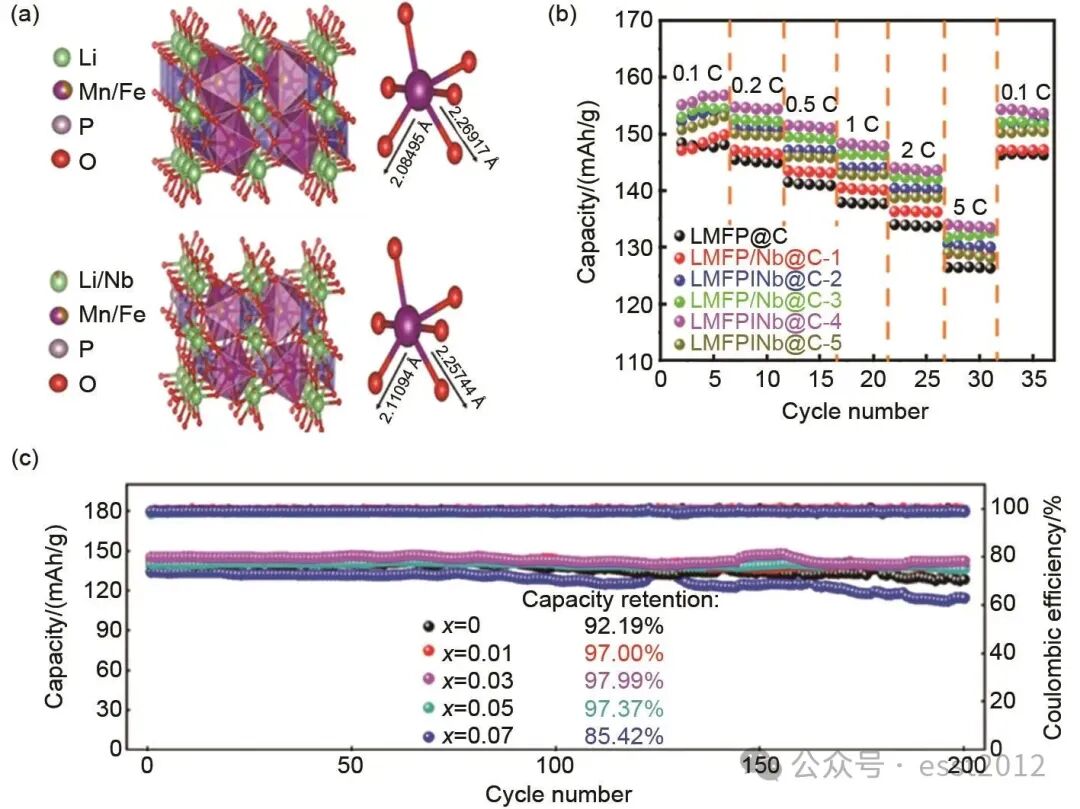
图9 (a) LFMP@C与LMFP/Nb@C的优化晶体结构及对应的MnO6八面体示意图;(b) LMFP/Nb@C倍率性能图;(c) Mg2+掺杂循环性能图
为系统对比不同掺杂策略对LMFP材料性能的调控效果,表2总结了具有代表性的单一及多元离子掺杂的研究数据。可以看出,无论是等价掺杂(如Ni2+、Mg2+)还是高价掺杂(如Ti4+、Nb5+),均能通过稳定晶体结构,优化离子传输动力学,有效降低界面阻抗、提高锂离子扩散系数,最终显著改善材料的可逆容量与倍率性能。例如,Lyu等通过系统调控Mn2+-Fe2+-Mg2+-C2O42--H2O体系的共沉淀行为,成功合成了成分均匀的(Fe0.4Mn0.6)1-xMgxC2O4前驱体。原子尺度的均匀掺杂不仅增强了结构稳定性,同时优化了离子传输动力学,如图9(c)所示,与未掺杂材料相比,显著提升了循环性能:2 C倍率下放电容量为144.04 mAh/g,200次循环后容量保持率高达97.99%,因为Mg2+掺杂促进Li+的传输,有助于减少极化现象。同时,Mg2+存在于晶格中不参与氧化还原反应,且其离子半径适中[r(Mn2+)>r(Fe2+)>r(Mg2+)>r(Mn3+)>r(Fe3+)],这有效缓解循环过程中的体积变化,维持材料的结构稳定性。共沉淀法能够实现掺杂元素在体相中的均匀分布,为制备高性能正极材料提供了可靠且具备规模化潜力的合成方案,展现了该方法在解决成分偏析与性能不均匀性方面的显著优势。
表2 不同元素掺杂对LiMnxFe1-xPO4/C性能的影响 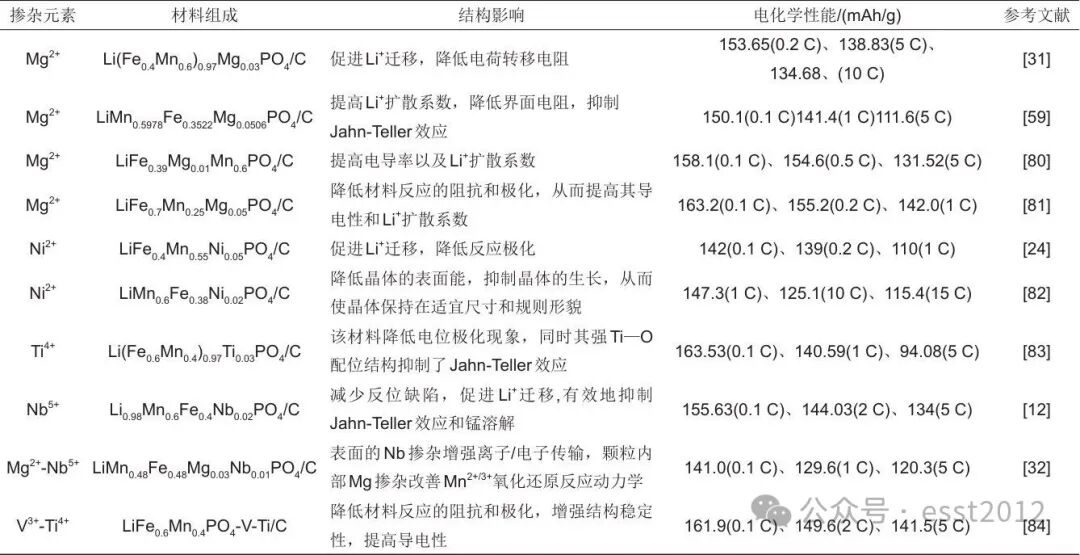
3 结论与展望
本文综述了共沉淀法在制备磷酸锰铁锂(LMFP)正极材料领域的研究进展。研究表明,该方法通过实现原子级别的均匀混合与组分的精确控制,能够有效提升前驱体的结构一致性,并优化LMFP材料的电化学性能。通过将共沉淀法与其他多种策略——如核壳结构与浓度梯度设计、碳包覆以及离子掺杂相结合,可进一步改善材料在电子导电性、锂离子扩散能力和结构稳定性方面的表现,从而为其工业化可控合成提供有效途径。尽管共沉淀法在前驱体组分均匀性与结构调控方面具有显著优势,LMFP材料仍面临本征电导率低、Jahn-Teller效应及其引发的锰溶出等问题。因此,未来研究应重点关注反应机理的深入解析、工艺参数的系统优化,以及多策略改性手段的有效整合。
为实现LMFP的大规模商业化应用,未来研究需致力于共沉淀法在工业级制备中的标准化与工艺优化,并可重点借鉴成熟LFP材料的产业化路径与技术经验,以开发稳定、可重复的前驱体合成路线。此外,还需进一步探究碳包覆、离子掺杂及梯度结构等多种策略的协同效应,并系统整合这些改性手段,以协同改善电子/离子电导率、抑制Jahn-Teller畸变与锰溶出现象,同时优化界面稳定性与锂离子传输动力学,从而显著提升材料的倍率性能与循环稳定性。
综上所述,磷酸锰铁锂(LMFP)材料凭借其高安全性和高能量密度优势,已成为下一代锂离子电池正极材料的理想候选者之一。为实现LMFP基电池的全面商业化应用,需进一步推动基础研究与产业化实践的深度融合,并持续优化材料设计与合成工艺,以期开发出兼具高性能与低成本优势的LMFP电池系统,从而推动其在大规模储能领域的广泛应用。
第一作者:贾雨(2000—),女,硕士研究生,研究方向为电化学储能器件以及关键材料;
通讯作者:屈龙,副教授,研究方向为电化学储能器件以及关键材料。
免责声明:本站所有信息均来源于互联网搜集,并不代表本站观点,本站不对其真实合法性负责。如有信息侵犯了您的权益,请告知,本站将立刻删除。

